Genetic variation of putative myokine signaling is dominated by biological sex and sex hormones
- LeandroMVelez12
- CassandraVan13
- TimothyMoore4
- ZhenqiZhou5
- CaseyJohnson13
- AndreaLHevener[email protected]56
- MarcusMSeldin[email protected]13
- Short Report
- Biochemistry and Chemical Biology
- Computational and Systems Biology
- myokine
- endocrinology
- systems genetics
- physiology
- Human
- publisher-id76887
- doi10.7554/eLife.76887
- elocation-ide76887
Abstract
Skeletal muscle plays an integral role in coordinating physiological homeostasis, where signaling to other tissues via myokines allows for coordination of complex processes. Here, we aimed to leverage natural genetic correlation structure of gene expression both within and across tissues to understand how muscle interacts with metabolic tissues. Specifically, we performed a survey of genetic correlations focused on myokine gene regulation, muscle cell composition, cross-tissue signaling, and interactions with genetic sex in humans. While expression levels of a majority of myokines and cell proportions within skeletal muscle showed little relative differences between males and females, nearly all significant cross-tissue enrichments operated in a sex-specific or hormone-dependent fashion; in particular, with estradiol. These sex- and hormone-specific effects were consistent across key metabolic tissues: liver, pancreas, hypothalamus, intestine, heart, visceral, and subcutaneous adipose tissue. To characterize the role of estradiol receptor signaling on myokine expression, we generated male and female mice which lack estrogen receptor α specifically in skeletal muscle (MERKO) and integrated with human data. These analyses highlighted potential mechanisms of sex-dependent myokine signaling conserved between species, such as myostatin enriched for divergent substrate utilization pathways between sexes. Several other putative sex-dependent mechanisms of myokine signaling were uncovered, such as muscle-derived tumor necrosis factor alpha (TNFA) enriched for stronger inflammatory signaling in females compared to males and GPX3 as a male-specific link between glycolytic fiber abundance and hepatic inflammation. Collectively, we provide a population genetics framework for inferring muscle signaling to metabolic tissues in humans. We further highlight sex and estradiol receptor signaling as critical variables when assaying myokine functions and how changes in cell composition are predicted to impact other metabolic organs.
Introduction
Proteins secreted from skeletal muscle, termed myokines, allow muscle to impact systemic physiology and disease. Myokines play critical roles in a variety of processes, including metabolic homeostasis, exercise improvements, inflammation, cancer, and cognitive functions 28Severinsen and Pedersen20205Eckel20196Febbraio and Pedersen20208Kim et al.20199Kim et al.202124Seldin and Wong2012. Several notable examples include key peptide hormones such as myostatin and interleukin-6 which exert potent actions in regulating autocrine/paracrine muscle physiology 10Kollias and McDermott2008 and beneficial exercise-induced endocrine signaling 28Severinsen and Pedersen2020, respectively. Despite the clear relevance of these factors in mediating a multitude of physiological outcomes, the genetic architecture, regulation, and functions of myokines remain inadequately understood. Given that genetic sex contributes critically to nearly every physiological outcome, it is essential to consider when relating specific mechanisms to complex genetic and metabolic interactions. Specifically, many metabolic traits impacted by myokines show striking sex differences arising from hormonal 18Mauvais-Jarvis et al.201717Mauvais-Jarvis20153Clegg and Mauvais-Jarvis201822Ribas et al.2016, genetic 18Mauvais-Jarvis et al.201734Zore et al.2018, or gene-by-sex interactions 20Norheim et al.20192Chella Krishnan et al.2021. In this study, we leveraged natural genetic correlation structure of gene expression both within and across tissues to understand how muscle interacts with metabolic tissues. Collectively, we provide a population genetics framework for inferring muscle signaling to metabolic tissues in humans. We further highlight sex and estradiol receptor signaling as critical variables when assaying myokine functions and how changes in cell composition are predicted to impact other metabolic organs.
Results
Sex hormone receptors are enriched with myokine expression independent of biological sex
Our goal was to exploit correlation structure of natural genetic variation to investigate how skeletal muscle communicates with and impacts metabolic organs. We first assayed regulation of myokines and changes in cellular composition, then related these observations to inferred cross-tissue signaling mechanisms (Figure 1A). Initially, we performed differential expression of genes encoding all known secreted proteins (3666 total) in skeletal muscle from 210 male and 100 female individuals 1Battle et al.2017. While several notable myokines appeared different between sexes (Figure 1B), a striking majority of all secreted proteins (74%) showed no difference in expression between males and females (Figure 1C, Supplementary file 1). To understand potential sex effects on the regulation of myokines, gene ontology (GO) enrichments were performed on genes which showed the strongest correlation with myokines corresponding to each category (male-specific, female-specific, or non-sex-specific). Here, the top 10 pathways which persisted in females were also always observed to overlap with the non-sex-specific category (Figure 1D). In contrast, pathways enriched for male-specific myokines were distinct (Figure 1D). Notably, the female and shared pathways suggested roles in epigenetics and RNA processing, while male-specific myokine coregulated processes were more enriched in metabolic pathways (e.g. NADH metabolism) (Figure 1D). Further, a majority of myokines showed strong correlation with receptors mediating functions of androgens (androgen receptor [AR]), estradiol (estrogen receptor α – ESR1), or both, regardless of sex-specific expression (Figure 1E). We note that expression of hormone receptors themselves were also not significantly different between sexes (Figure 1—figure supplement 1). To infer causality from hormone receptor regulation, we performed RNA-sequencing (RNA-Seq) on mice lacking Esr1 in skeletal muscle specifically (MERKO) and integrated these analyses with human myokine estimates. While myokines not regulated by Esr1 showed little sex-specific differences in expression, those which were estrogen-dependent showed much stronger representation of sex specificity, in particular in males (Figure 1F–G). Among these was the master regulator of skeletal muscle differentiation and proliferation, myostatin (MSTN), where hormone receptor correlations and gene expression were markedly higher in males compared to females (Figure 1H). Further, ablation of Esr1 in mice uniquely drove expression changes in males (Figure 1H). These data suggest interactions between biological sex and ESR1 to tightly regulate MSTN in males, where other factors could contribute more in females. Given that, like many bioactive secreted proteins, the regulation and sex specificity of myostatin are additionally controlled via post-transcriptional mechanisms 19McMahon et al.2003, we next explored gene expression changes at the protein level. Immunoblots were performed on skeletal muscle from male and female WT or MERKO mice (Figure 1—figure supplement 2). Quantification of the processed form of myostatin showed that, consistent with the RNA-Seq in mice and humans, the protein trended toward higher levels in male compared to female mice, where ablation of Esr1 showed a reduction (Figure 1I). Dissimilar to the mouse sequencing data but consistent with human correlations, female MERKO mice showed a reduction in processed form of myostatin relative to their WT controls (Figure 1I). Related to the sex-specific regulation of myostatin observed at both RNA and protein levels, gene expression also showed differences in functional annotations. Here, the most highly enriched pathways in males showed GO terms related to glycolytic metabolism (Figure 1J) compared to oxidative phosphorylation in females (Figure 1K). These observations are consistent with previous studies which note myostatin-dependent increases in muscle mass in males, but not females 19McMahon et al.200321Reisz-Porszasz et al.2003, where estradiol signaling is suggested as a mechanism mediating these differences. These data demonstrate that, expression of most myokines are not different between genetic sexes; however, interactions between sex and hormone receptors likely play important roles in determining myokine regulation and local signaling.
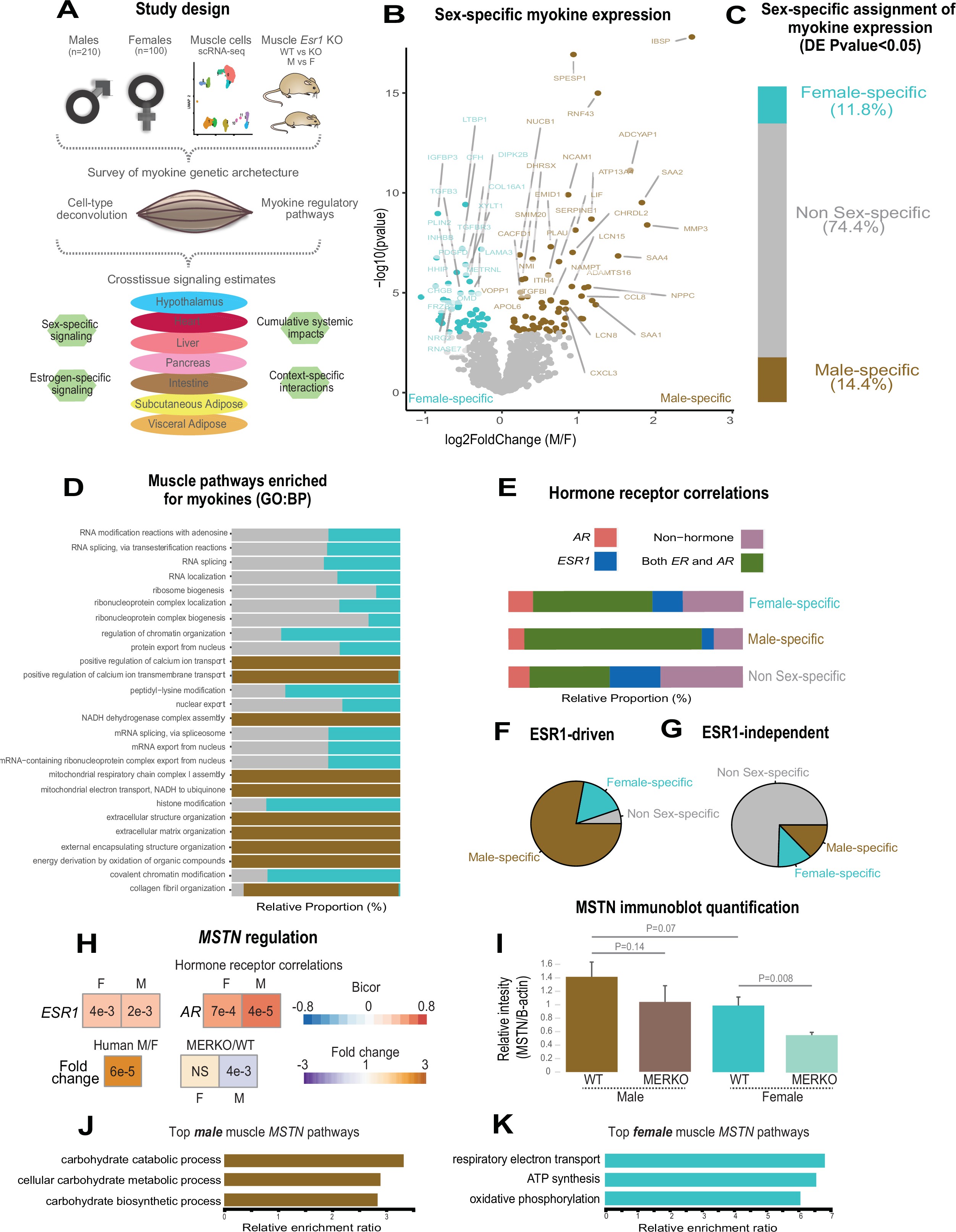
Sex and hormone effects on myokine regulation.
(A) Overall study design for integration of gene expression from muscle from 310 humans, single-cell RNA-sequencing (RNA-seq), muscle-specific deletion of Esr1 to infer interorgan coregulatory process across major metabolic tissues. (B–C) Differential expression analysis for sex was performed on all genes corresponding to secreted proteins in skeletal muscle (myokines). The specific genes which showed significant changes in each sex are shown as a volcano plot (B) and the relative proportions of myokines corresponding to each category at a least-stringent logistic regression p-value less than 0.05 (C). (D) For each differential expression category based on sex shown in C, myokines were correlated with all other muscle genes for pathway enrichment. Then the top 10 enriched pathways in males, females, or non-sex specific (by overall significance) were visualized together where number of genes corresponding to each category shown as a relative proportion. (E) The same analysis as in D, except instead of myokines being correlated with AR, ESR1, both hormone receptors, or neither, as compared to correlating with all genes. (F–G) Myokines were binned into two categories based on significant differential expression (logistic regression adjusted p-value < 0.05) between muscle-specific WT and MERKO mice (F) or those that showed no change (G), then visualized as relative proportions within each category shown in (C). (H) Midweight bicorrelation (bicor) coefficients (color scheme) and corresponding regression p-values (filled text) are shown for muscle MSTN ~ESR1 or AR in both sexes (top). Below, correlations are shown for differential expression log2FC (color scheme) and corresponding logistic regression p-values (text fill) for MSTN between sexes in humans or WT vs. MERKO mice. (I) Quantification of processed form of myostatin (Figure 1—figure supplement 2, bottom band) relative to β-actin in WT or MERKO muscle in male or female mice. p-Values calculated using a Student’s t-test. (J–K) The top three pathways of genes which significantly (p < 1e-4) correlated with muscle MSTN in males (J) or females (K). For human data, n = 210 males and n = 100 females. For mouse MERKO vs. WT comparisons, n = 3 mice per group per sex. p-Values from midweight bicorrelations were calculated using the Student’s p-value from WGCNA and logistic regression p-values were calculated using DESeq2.
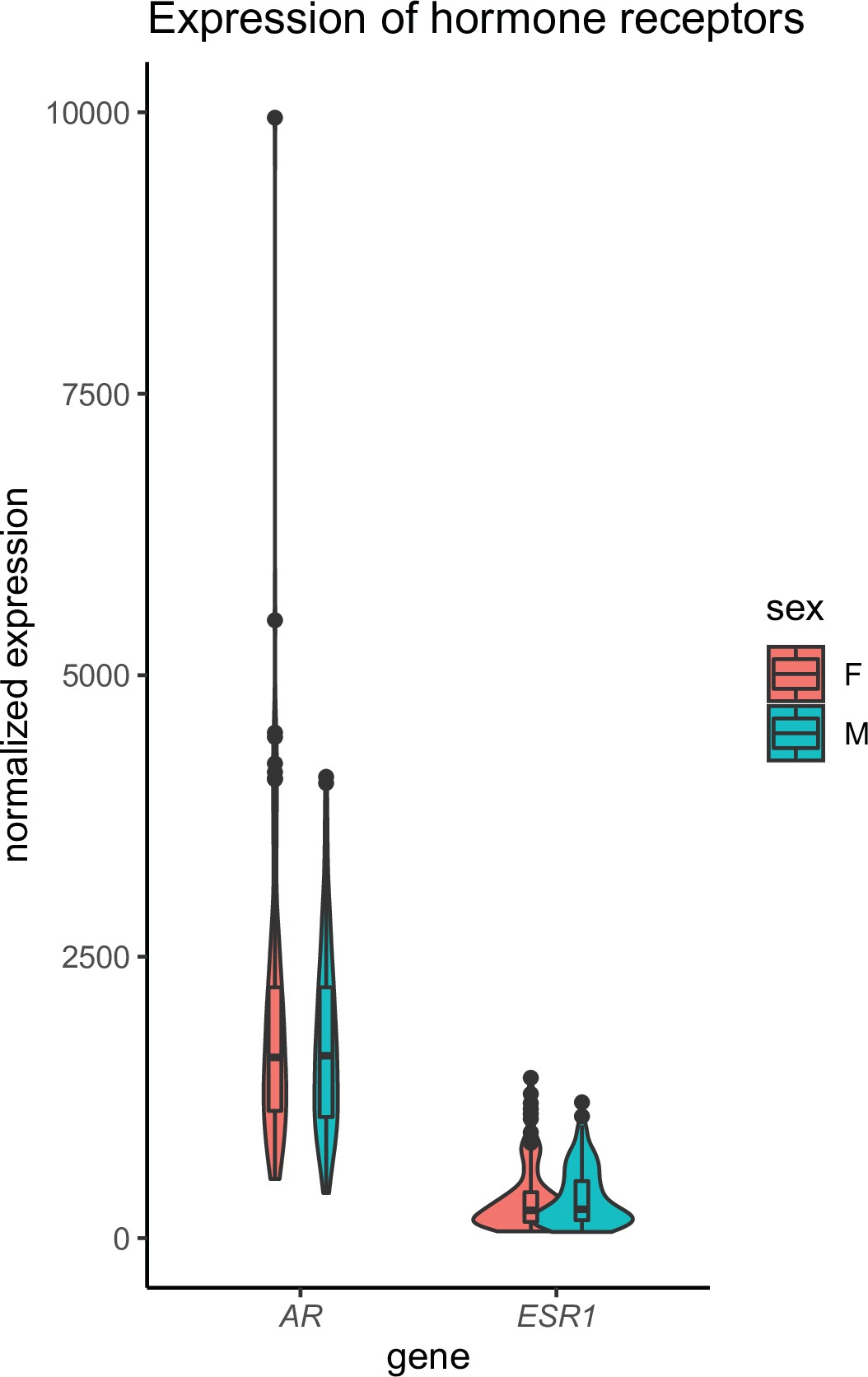
Skeletal muscle sex hormone receptor expression between sexes.
Normalized gene expression levels for androgen receptor (AR) or estrogen receptor (ESR1) (y-axis) in each sex (x-axis). None of the expression levels were significantly different between sexes (Student’s t-test).
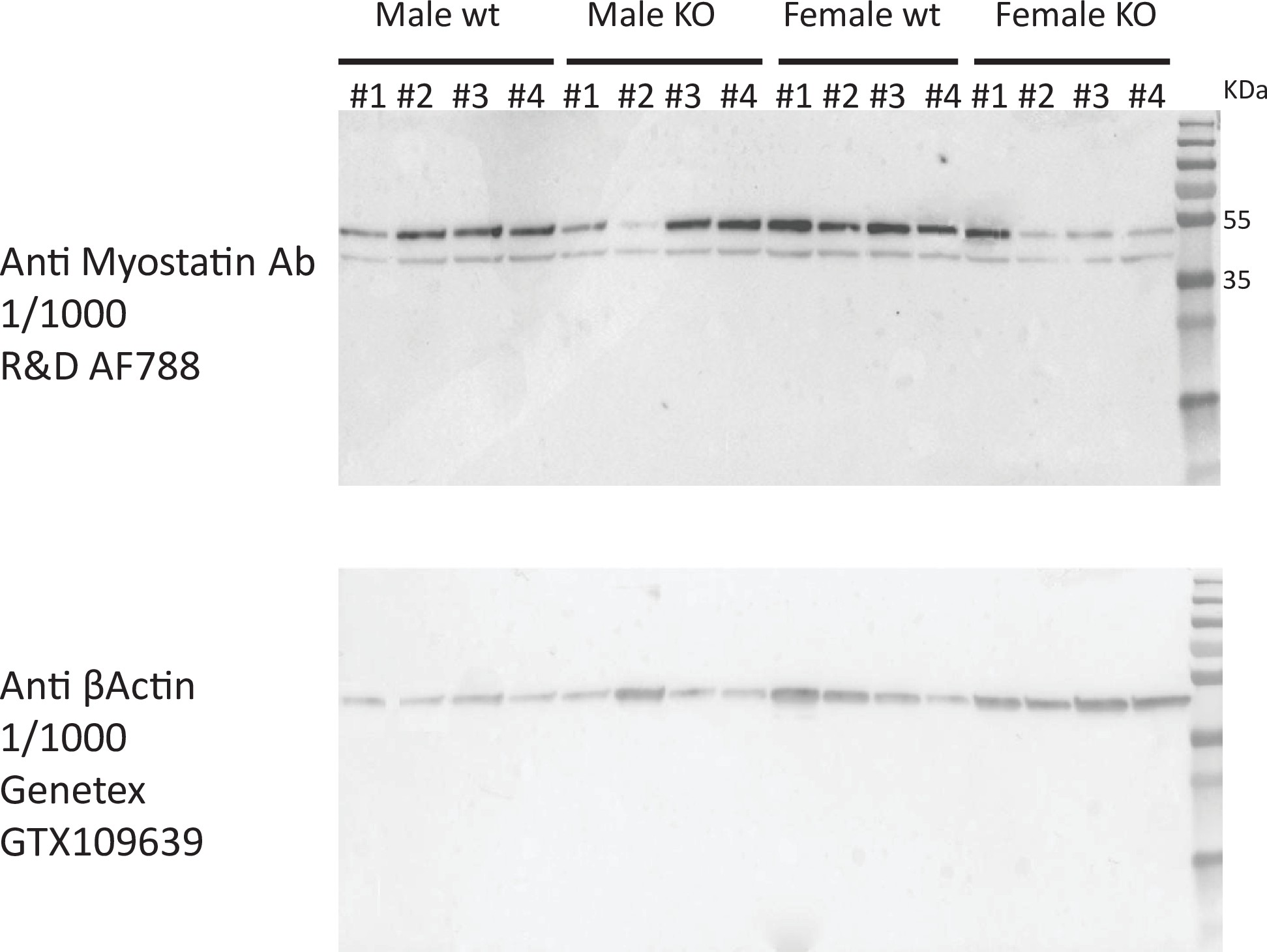
Immunoblot for myostatin in EDL muscle from WT and MERKO male and female mice.
Full immunoblots shown for skeletal muscle lysate blotted for myostatin (top) or β-actin (bottom) corresponding to different C57BL/6J male (left) or female (right) mice in either WT (floxed) or KO (floxed-cre) for skeletal muscle Esr1. Band sizes shown to indicate either precursor (top band) or processed/LAP form (bottom band) of myostatin.
Sex dominates cross-tissue pathways enriched for myokines
Given that expression levels of most myokines appeared similar between sexes, we next assessed putative functions across organs. We applied a statistical method developed to infer cross-tissue signaling which occur as a result of genetic variation 25Seldin et al.201826Seldin and Lusis201927Seldin et al.2019. Here, we assayed the distribution of midweight bicorrelation coefficients between myokine expression levels and global gene expression in key metabolic tissues including hypothalamus, heart, intestine, pancreas, liver, subcutaneous, and visceral adipose tissue. Remarkably, nearly all highly significant correlations between myokines and target organ genes (putative direct interactions) showed sex-specific modes of operation (Figure 2A–H). This sex specificity also appeared more pronounced for positive correlations between myokines and target tissue genes, as compared to negative (Figure 2H). Further, among these significant cross-tissue circuits, hormone receptor enrichments for these myokines were strongly dependent on the category (e.g. significant only in females) rather than target tissue (Figure 2A–H). This observation further suggests that hormone receptor levels (ESR1 or AR) in muscle are a stronger determinant of myokine expression compared to genetic sex; however, sex is suggested to dominate coregulated signaling processes across organs via myokines. To gauge the relative impact of muscle steroid hormone receptors across organs, the number of significant correlations between ESR1, AR, or both were quantified from muscle to each tissue. Here, ESR1 showed an order of magnitude stronger enrichment across metabolic tissues compared to AR or correlation with both hormone receptors (Figure 2I–J). Additionally, the number of significantly correlated cross-tissue male ESR1 genes (Figure 2I) was threefold higher than females (Figure 2J). Because both sex and ESR1 signaling appeared to contribute to the regulation and functions of myokines, significant cross-tissue enrichments were binned into categories taking into consideration whether myokines were driven by ESR1 in muscle, and/or showing a sex-specific mode of cross-organ significance. This analysis suggested that a majority of myokines were either driven by ESR1 and signaled robustly across sexes (Figure 2K, yellow) or signaled differently between sexes, but regulated independent of ESR1 (Figure 2K, red). These categories appeared to a much greater extent compared to a combination of both ESR1-driven myokine and sex-specific cross-tissue signaling (Figure 2K, beige) or neither (Figure 2K, seagreen). One notable example of predicted sex-specific signaling was observed for tumor necrosis factor alpha (TNFA). When compared between sexes, muscle TNFA showed markedly different putative target tissues (Figure 2L, left), as well as underlying functional pathways (Figure 2L, right). For example, overall inflammatory processes engaged by TNFA were stronger in adipose tissue in females; however, the same pathways were higher in liver and hypothalamus in males (Figure 2L, left). Collectively, these data show that genetic sex and related sex steroid hormones, particularly estradiol, exert dominant roles in regulating predicted tissue and pathway engagement by myokines.
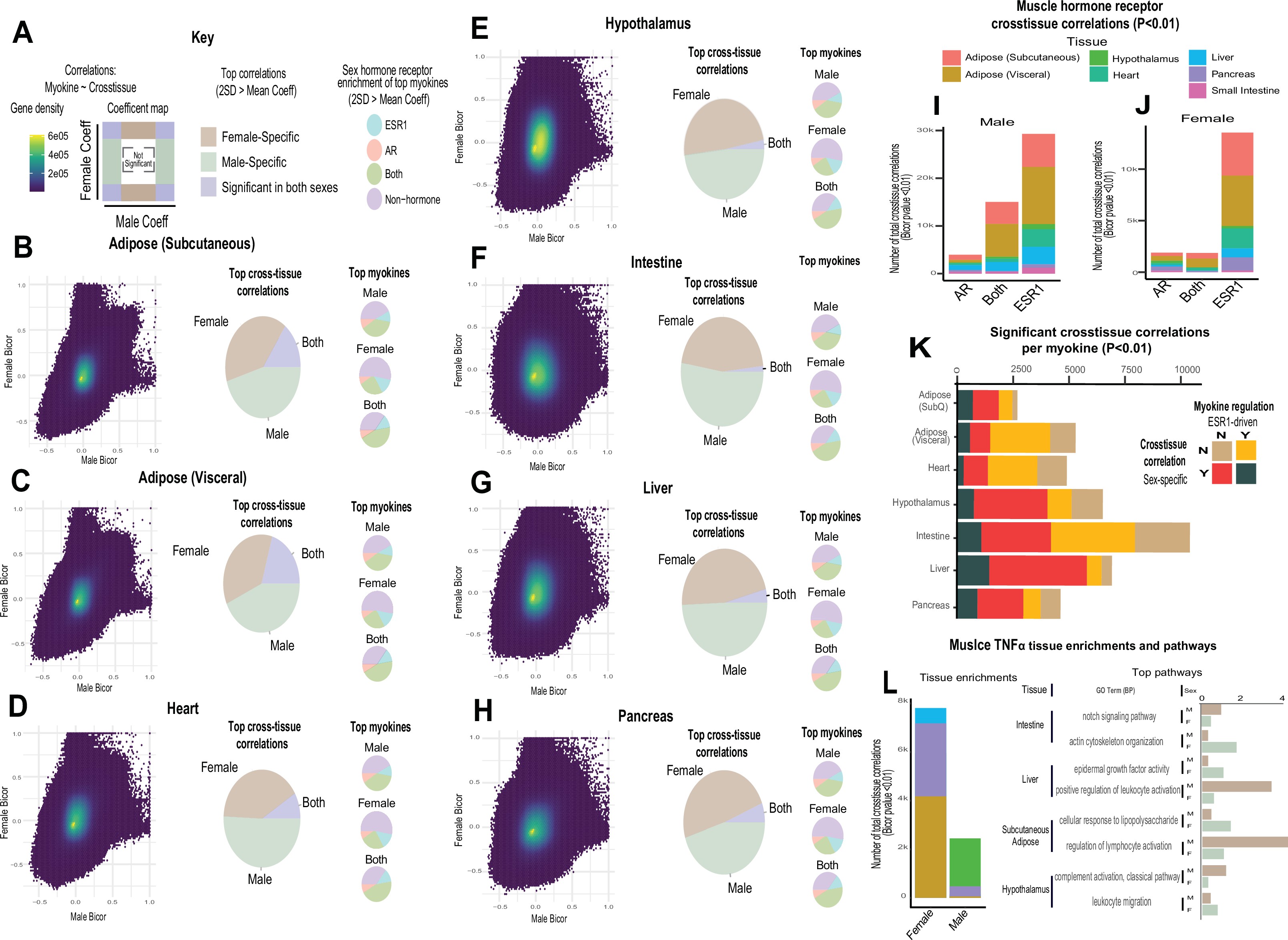
Sex and hormone effects on myokine regulation.
(A–H) Key illustrating analysis for distribution of midweight bicorrelation coefficients between all myokines in skeletal muscle and global transcriptome measures in each target tissue. Coefficients are plotted between sexes (left), where proportions for 2SD > mean are subdivided into occurrence uniquely in females, males, or shared (middle). The significant (2SD > mean) myokines identified in each category were then binned into hormone receptor correlations for ESR1, AR, both, or neither (right). This analysis was performed on all myokines across subcutaneous adipose tissue (B), visceral adipose (C), heart (D), hypothalamus (E), small intestine (F), liver (G), and pancreas (H). (I–J) Significant cross-tissue correlations between muscle ESR1, AR, or both hormone receptors are colored by tissue and shown for males (I) or females (J). (K) For each tissue (y-axis), the ratio of significant cross-tissue correlations per muscle myokine (x-axis) are shown and colored by categories of either the myokine regulated by ESR1 and/or a significant target tissue regression occurring specifically in one sex. (L) Number of significant cross-tissue correlations with muscle TNFα are shown for each sex and colored by tissue as in I–L (left). The −log10(p-value) of significance in an overrepresentation test (x-axis) are shown for top significant inter-tissue pathways for muscle TNFα in each sex (right).
Muscle cell proportions are similar between sexes, but associated changes across tissues show sex specificity
To determine the potential impact of muscle composition on other tissues, we next surveyed muscle cellular proportions in the context of genetics and sex. Single-cell sequencing of human skeletal muscle 23Rubenstein et al.2020 was integrated using cellular deconvolution 4Danziger et al.2019 to roughly estimate cellular composition in the population (Figure 3A). Here, a proportion in admixture approach 12Langfelder and Horvath2008 outperformed other methods (Figure 3—figure supplement 1) to capture a majority of established cell populations across individuals (Supplementary file 2). Similar to myokine expression, no notable differences were observed between sexes in terms of cell composition, with the exception of modest higher glycolytic fiber in males, compared to elevated oxidative fiber levels in females (Figure 3B). Additionally, no differences were observed in the correlations within muscle between compositions (Figure 3—figure supplement 2); however, nearly every cross-tissue enrichment corresponding to an individual muscle cell type differed between sexes (Figure 3C). Generally, differences in skeletal muscle cell abundance was associated with changes in liver and visceral adipose tissue pathways in males, compared to pancreas in females (Figure 3C). In contrast to general myokine enrichments, cell proportions showed stronger correlations with AR when compared to ESR1 across both sexes where the most abundant cell types were significantly enriched for both steroid hormone receptors (Figure 3D). To uncover potential direct mechanisms linking changes in cell composition to peripheral tissues, we correlated all myokines with cell composition profiles. Again, despite few differences between sexes in terms of myokine expression and cell composition, specific myokines highly correlating with individual cell types were markedly different between males and females with the exception of one, APOD in slow-twitch fibers (Figure 3E). To determine if variation in cell compositions corresponding to sex-specific tissue signaling via myokines was predicted to be likely, we implemented adjusted regression mediation analyses 33Yokota et al.2020 for glycolytic fiber composition. Because male glycolytic fiber-type abundance was selectively enriched for liver pathways such as immune cell activation and regulated exocytosis (Figure 3F), the top genes driving these enrichments were used for mediation. The top-correlated muscle secreted protein with male glycolytic fiber-type levels was secreted glutathione peroxidase 3 (GPX3). Here, adjusting regressions between glycolytic fiber and liver pathways on GPX3 reduced the overall significance across tissues (Figure 3G), suggesting GPX3 as a potential mediator of this communication. These data point to a potential mechanism whereby muscle fiber abundance could buffer free radical generation in the liver, thereby feeding back on inflammation. This analysis appeared additionally sensitive to inferring non-dependent relationships between muscle cell types, top-ranked myokines, and cross-tissue processes. For example, female glycolytic fibers were strongly enriched for pancreatic protein synthesis pathways; however, when adjusted for the top-ranked myokine CES4A, no changes in regression significance were observed (Figure 3F–G). These analyses suggest that male GPX3 is a potential mechanism whereby fast-twitch muscle signals to liver; however, the same cell type in females are predicted to drive pancreas protein synthesis independent of CES4A. In summary, we show that cell composition is strongly conserved between sexes, but putative cross-tissue signaling of altered composition differs entirely. We further suggest putative myokines and mechanisms, as well as highlight the key regulatory roles of estrodiol in both sexes.
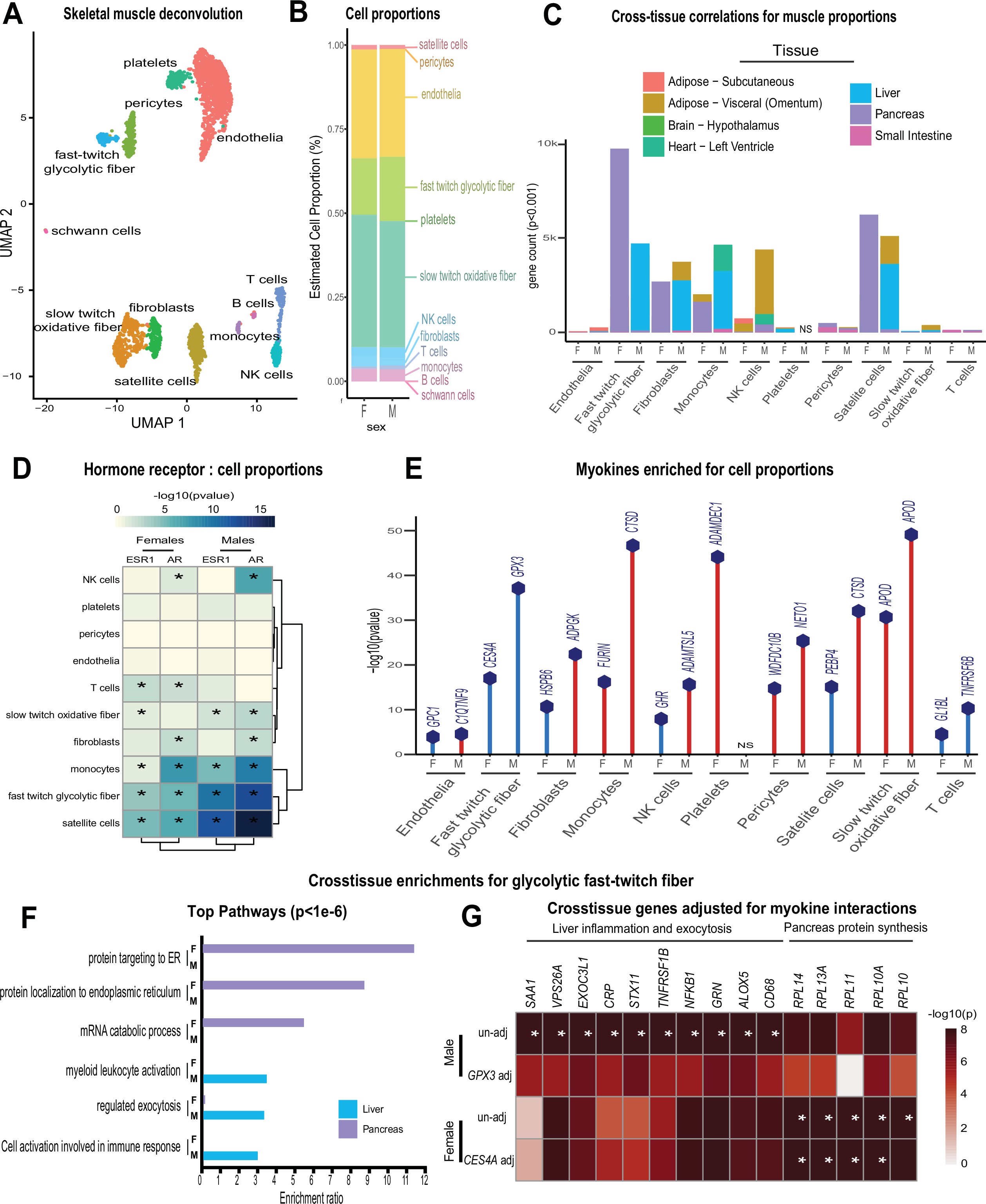
Genetic variation of muscle cell proportions and coregulated cross-tissue processes.
(A) Uniform Manifold Approximation and Projection (UMAP) for skeletal muscle single-cell sequencing used to deconvolute proportions. (B) Mean relative proportions of pseudo-single-cell muscle cell compositions (denoted by color) between sexes. (C) Number of significant cross-tissue correlations (y-axis) corresponding to each skeletal muscle type in each sex (x-axis). Target tissues are distinguished by color, where NS (male platelets) denotes that no significant cross-tissue correlations were observed. (D) Heatmap showing significance of correlations between skeletal muscle hormone receptors and cell proportions, * = p < 0.01. (E) The strongest enriched myokines are plotted for each myokine (y-axis, −log10 p-value of myokine ~ cell composition) are shown for each muscle proportion for each sex (x-axis). Gene symbols for myokines are shown above each line, where red lines indicate positive correlations between myokine and cell type and blue shows inverse relationships. (F) Significant cross-tissue correlated genes in liver (blue) and pancreas (purple) for muscle fast-twitch glycolytic fibers (p < 1e-6) were used for overrepresentation tests where enrichment ratio of significance (x-axis) is shown for each pathway and sex (y-axis). (G) Heatmap showing the regression significance of the top five genes corresponding to inflammation (liver), exocytosis (liver), and protein synthesis (pancreas) for proportions of fast-twitch fiber type (un-adj). Below each correlation between fast-twitch fiber and liver or pancreas gene, the same regressions were performed while adjusting for abundance of select myokines in each sex. * = p < 1e-6.
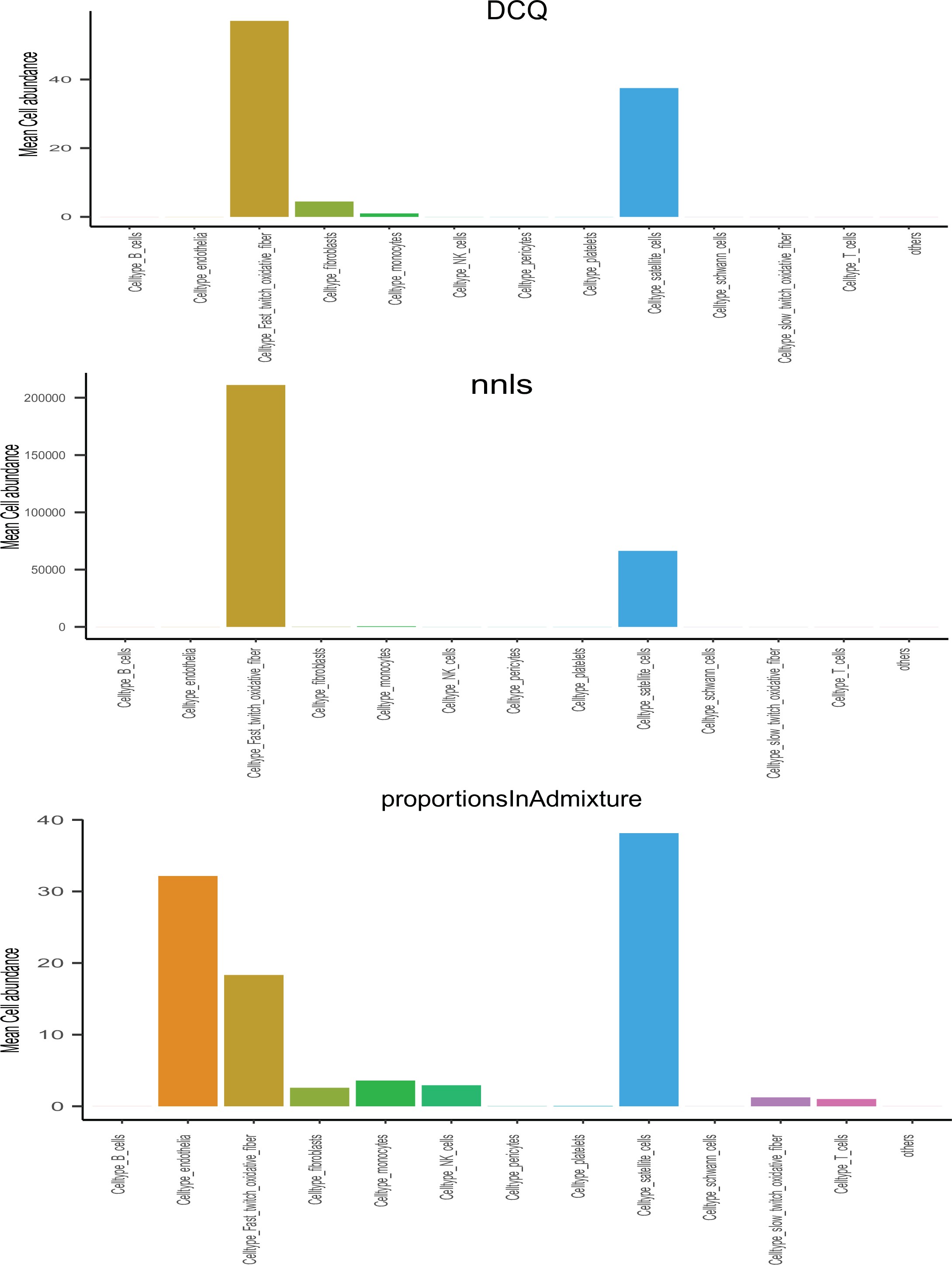
Comparisons of deconvolution methods.
Cell proportions were estimated from skeletal muscle sequencing across the 310 individuals in GTEx.
Here, comparisons of the three most common methods (DCQ, NNLS, and porportionsInAdmixture) were plotted for each pseudo-sc-proportion, where proportionsInAdmixture method captured the largest relative number of cell types.
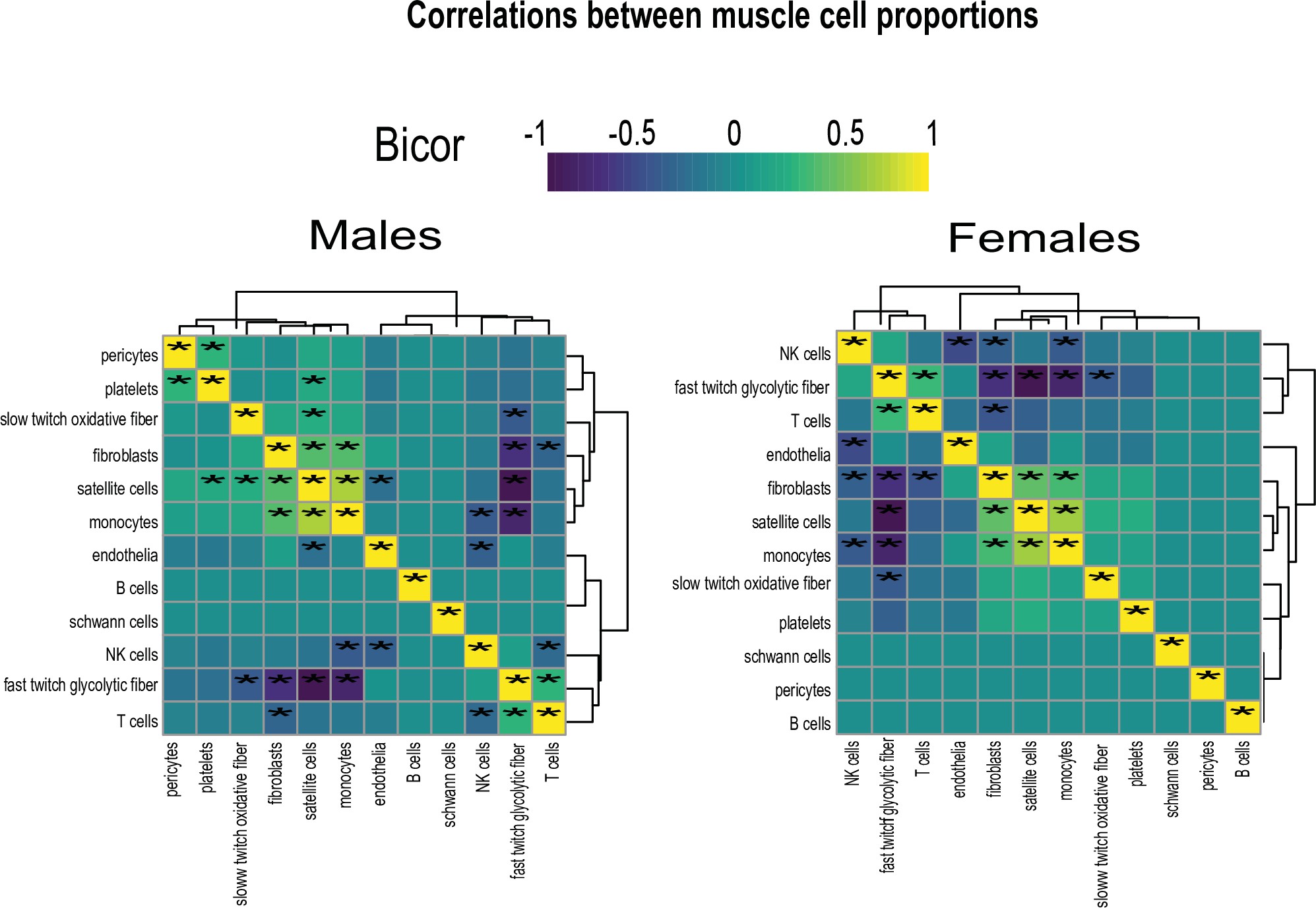
Cell composition correlations within each sex.
Heatmaps showing regressions for cell proportions in males (left) or females (right), * = regression p-value < 0.01.
Discussion
Here, we provide a population survey of skeletal muscle myokine regulation and putative functions using genetic variation and multi-tissue gene expression data. We find that in general, expression of myokines do not significantly differ between sexes; however, inferred signaling mechanisms across tissues using regressions show strong sex specificity. Steroid hormone receptors, in particular ESR1, is highlighted as a key regulator of myokines and potentially interacting with biological sex for proteins such as myostatin. Further integration with loss-of-function mouse models of Esr1 highlighted the key roles of estradiol signaling in muscle in terms of myokine regulation and signaling across both sexes. Generation of pseudo-single-cell maps of muscle composition showed that, like myokines, muscle proportions are conserved between sexes, but inferred interorgan consequences differ substantially. When interpreting these findings, several considerations should be taken. While inter-tissue regression analyses have been informative to dissect mechanisms of endocrinology 25Seldin et al.201826Seldin and Lusis201927Seldin et al.201913Li and Auwerx2020, observations can be subjected to spurious or latent relationships in the data. While causality for inter-organ signaling can be inferred statistically using approaches such as mediation as in Figure 3H, the only methods to provide definitive validation for new mechanisms are in experimental settings. Further, our current analyses rely on gene expression to guide functions of proteins which are typically strongly regulated by post-transcriptional processes. As shown for myostatin (Figure 1), gene expression analyses can miss key functional aspects of proteins, where follow-up studies and resources focused on protein and subsequent modification levels could heavily improve predictions. In addition, we anticipate that estimates for ESR1 effects on myokines in this study likely represent an underestimated number of all human ESR1-driven myokines. One limitation here includes that annotation of known orthologous mouse-human genes 29The Alliance of Genome Resources Consortium2020 remains somewhat limited. Furthermore, cell composition estimates from single-cell sequencing data are inferred from gene expression, where histological or flow cytometry-based methods can provide much more accurate direct quantifications. Clearly, morphological and structural differences between sexes have been observed in humans 7Haizlip et al.2015 which, if not apparent in deconvoluted gene expression, would be missed in this analysis. Future studies addressing these points will help to clarify context- and mechanism-relevant muscle-derived endocrine communication axes. In summary, this study highlights the key contributions of sex and sex steroid hormones in mediating myokine functions.
Materials and methods
All datasets used, R scripts implemented for analyses, and detailed walkthrough guide is available via: https://github.com/Leandromvelez/myokine-signaling, (copy archived at swh:1:rev:530dc39df1c586ad67eab36688fa2c1936b06354; 31Velez and Seldin2022).
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Antibody | Anti-MSTN(Goat polyclonal) | R&D | AF788 | (1:1000) |
| Antibody | Rabbit anti-βactin(Rabbit polyclonal) | Genetex | GTX109639 | (1:1000) |
Animals
All mice used in this study were approved by the University of California Los Angeles (UCLA) Animal Care and Use Committee, in accordance with Public Health Service guidelines with reference #92-169.
Data sources and availability
All data used in this study can be immediately accessed via GitHub to facilitate analysis. Human skeletal muscle and metabolic tissue data were accessed through GTEx V8 downloads portal on August 18, 2021, and previously described 1Battle et al.2017. To enable sufficient integration and cross-tissue analyses, these data were filtered to retain genes which were detected across tissues where individuals were required to show counts >0 in 1.2e6 gene-tissue combinations across all data. Given that our goal was to look across tissues at enrichments, this was done to limit spurious influence of genes only expressed in specific tissues in specific individuals. Post-filtering consists of 310 individuals and 1.8e7 gene-tissue combinations. Single-cell sequencing from skeletal muscle used for deconvolution was obtained from 23Rubenstein et al.2020. Esr1 WT and KO mouse differential expression results are available on GitHub as well, where raw sequencing data has been deposited in NIH sequence read archive (SRA) under the project accession: PRJNA785746.
Selection of secreted proteins
To determine which genes encode proteins known to be secreted as myokines, gene lists were accessed from the Universal Protein Resource which has compiled literature annotations terms for secretion 30The UniProt Consortium2021. Specifically, the query terms to access these lists were: locations:(location:"Secreted [SL-0243]" type:component) AND organism:"Homo sapiens (Human) 9Kim et al.2021ref606" where 3666 total entries were found.
Differential expression of myokines dependent on sex
Gene expression counts matrices were isolated from the rest of the tissues, where individual genes were retained if the total number of counts exceeded 10 in 50 individuals. Next, only genes encoding secreted proteins (above) were retained, where logistic regression contrasted on sex was performed using DESeq2. Differential expression summary statistics were used for downstream binning of sex specificity based on an empirical logistic regression p-value < 0.05. This threshold was used to reflect a least stringent cutoff where, despite potential false positive influence, genes which nominally trended toward sex-specific expression could be included in those categories. Given that the general conclusions supported very few proportions of myokines showing sex-specific patterns of expression, this conclusion would only be further exaggerated if the differential expression threshold were made more stringent and lessened the number of myokines in each category.
Regression analyses across tissues
Regression coefficients and corresponding p-values across tissues were generated using WGCNA bicorandpvalue() function 12Langfelder and Horvath2008. Myokine-target gene pairs were considered significant (e.g. Figure 2A–H) at a threshold of abs(bicor) > 2 standard deviations beyond the average coefficient for the given target tissue of interest. In previous studies, this threshold of 2 standard deviations reflects adaptive permutation testing p-values < 0.01 25Seldin et al.201826Seldin and Lusis2019. For analyses estimating cumulative patterns of concordance across tissues (e.g. Figure 2I–L), empirical regression p-values (Student’s p-value from bicor coefficients) of 0.01 (corresponding to abs(bicor) > 0.1) were used to assay global patterns. While usage of empirical p-values are clearly subjected to false positives, these were used for several analyses to capture broad visualization of both potential direct interactions which would show significance across multiple FDR thresholds (e.g. myokine-target gene), as well as coregulated indirect processes across organs. Thus, assessing cumulative changes as a result of larger physiologic shifts. It is important to note that we exclusively rely on these empirical p-values when surveying broad correlation structures, whereas much more stringent and appropriate thresholds (e.g. p < 1e-6 for Figure 3G) were applied when inferring direct interactions.
Pathway enrichment analyses
For Figures 1I and 3G, genes corresponding to p-value cutoffs were visualized using Webgestalt 15Liao et al.2019 to enable streamline analysis. This tool enabled simultaneous overrepresentation testing of GO:BP (non-redundant), KEGG, and Panther databases. For Figure 1D, the top 1000 (by regression p-value) significant genes from myokines to all muscle bicorrelation analysis in females, males, or non-sex specific datasets were assessed for enrichment in GO Biological Process terms using ClusterProfiler v4.0.2 in R 32Wu et al.2021. The resulting top 10 GO terms in each dataset were integrated and plotted against the relative proportion of the p-adjusted value and visualized in the same graph using ggplot2.
Deconvolution of skeletal muscle
Raw single-cell RNA-sequencing from skeletal muscle was obtained from 23Rubenstein et al.2020. These raw counts were analyzed in Seurat where cluster analyses identified variable cell compositions. Cell type annotations were assigned based on the top 30 genes (Supplementary file 2) assigned to each Uniform Manifold Approximation and Projection (UMAP) cluster through manual inspection and ENRICHR 11Kuleshov et al.2016. Finally, a normalized matrix of gene:cells was exported from Seurat and used to run deconvolution on skeletal muscle bulk sequencing. Using the ADAPTS pipeline 4Danziger et al.2019, three deconvolution methods (nnls, dcq, or proportions in admixture) were compared based on ability to robustly capture cell proportions (Figure 3—figure supplement 1), where proportion in admixture showed the best performance and subsequently applied to bulk sequencing.
ESR1 muscle KO generation, RNA-Seq, and integration with human data
Muscle-specific Esr1 deletion was generated and characterized as previously described 22Ribas et al.2016. Whole quadriceps was pulverized at the temperature of liquid nitrogen. Tissue was homogenized in Trizol (Invitrogen, Carlsbad, CA), RNA was isolated using the RNeasy Isolation Kit (Qiagen, Hilden, Germany), and then tested for concentration and quality with samples where RIN > 7.0 used in downstream applications. Libraries were prepared using KAPA mRNA HyperPrep Kits and KAPA Dual Index Adapters (Roche, Basel, Switzerland) per manufacturer’s instructions. A total of 800–1000 ng of RNA was used for library preparation with settings 200–300 bp and 12 PCR cycles. The resultant libraries were tested for quality. Individual libraries were pooled and sequenced using a HiSeq 3000 at the UCLA Technology Center for Genomics and Bioinformatics (TCGB) following in-house established protocols. Raw RNA-Seq reads were inspected for quality using FastQC v0.11.9 (Barbraham Institute, Barbraham, England). Reads were aligned and counted using the Rsubread v2.0.0 14Liao et al.2019 package in R v3.6 against the Ensembl mouse transcriptome (v97) to obtain counts. Lowly expressed genes (>80% samples with 0 count for particular gene) were removed. Samples were analyzed for differential expression using DeSeq2 v1.28.0 16Love et al.2014.
Conservation of gene between mice and humans
To find which myokines and pathways were conserved between mice and humans, all orthologous genes were accessed from MGI vertebrate homology datasets, which have been compiled from the Alliance for Genome Resources 29The Alliance of Genome Resources Consortium2020 and intersected at the gene level (roughly 18,000 genes).
Immunoblotting procedures
Muscle tissue was homogenized in the TissueLyser II (Qiagen) at 4 °C in RIPA lysis buffer supplemented with protease inhibitors. The homogenate was centrifuged at 4 °C for 10 min at 10,000 g, and the protein concentrations in the supernatant were measured by the BCA assay (Bio-Rad). After boiling protein samples for 5 min, 20 μg of protein from each sample were applied on an SDS-polyacrylamide gel (10%) and electrophoresis was performed at 100 V for 1.5 hr. The separated proteins were transferred to nitrocellulose membranes and membranes were blocked for 1.5 hr in TBS (4 mM Tris-HCl, pH 7.5, and 100 mM NaCl) containing 5% skim milk plus Tween 20, at room temperature. Goat polyclonal anti-GDF8 (Myostatin) (R&D, catalog number AF788) at 1/1000 dilution were applied overnight as primary antibody. After washings, membranes were incubated with Goat IgG HRP-conjugated Antibody (R&D HAF017) at 1/10,000 for 2 hr, and bound HRP activity was detected with an enhanced chemiluminescence method (Clarity Western ECL, Bio-Rad), by means of a chemiluminescence detection system (ChemiDoc System, Bio-Rad). The intensities of the resulting bands were quantified by densitometry (ImageJ free software). Membranes were immersed in a stripping solution for 10 min (Restore PLUS Western Blot, Thermo Fisher), and then the process repeated with a rabbit polyclonal anti-β-actin (GeneTex GTX109639) at 1/1000 dilution as loading control to assess uniformity of loading.
References
-
- ABattle
- CDBrown
- BEEngelhardt
- SBMontgomery
-
- KChella Krishnan
- LVergnes
- RAcín-Pérez
- LStiles
- MShum
- LMa
- EMouisel
- CPan
- TMMoore
- MPéterfy
- CERomanoski
- KReue
- JLMBjörkegren
- MLaakso
- MLiesa
- AJLusis
-
- DJClegg
- FMauvais-Jarvis
-
- SADanziger
- DLGibbs
- IShmulevich
- MMcConnell
- MWBTrotter
- FSchmitz
- DJReiss
- AVRatushny
-
- JEckel
-
- MAFebbraio
- BKPedersen
-
- KMHaizlip
- BCHarrison
- LALeinwand
-
- SKim
- J-YChoi
- SMoon
- D-HPark
- H-BKwak
- J-HKang
-
- JSKim
- DAGalvão
- RUNewton
- EGray
- DRTaaffe
-
- HDKollias
- JCMcDermott
-
- MVKuleshov
- MRJones
- ADRouillard
- NFFernandez
- QDuan
- ZWang
- SKoplev
- SLJenkins
- KMJagodnik
- ALachmann
- MGMcDermott
- CDMonteiro
- GWGundersen
- AMa’ayan
-
- PLangfelder
- SHorvath
-
- HLi
- JAuwerx
-
- YLiao
- GKSmyth
- WShi
-
- YLiao
- JWang
- EJJaehnig
- ZShi
- BZhang
-
- MILove
- WHuber
- SAnders
-
- FMauvais-Jarvis
-
- FMauvais-Jarvis
- APArnold
- KReue
-
- CDMcMahon
- LPopovic
- FJeanplong
- JMOldham
- SPKirk
- CCOsepchook
- KWYWong
- MSharma
- RKambadur
- JJBass
-
- FNorheim
- YHasin-Brumshtein
- LVergnes
- KChella Krishnan
- CPan
- MMSeldin
- STHui
- MMehrabian
- ZZhou
- SGupta
- BWParks
- AWalch
- KReue
- SMHofmann
- APArnold
- AJLusis
-
- SReisz-Porszasz
- SBhasin
- JNArtaza
- RShen
- ISinha-Hikim
- AHogue
- TJFielder
- NFGonzalez-Cadavid
-
- VRibas
- BGDrew
- ZZhou
- JPhun
- NYKalajian
- TSoleymani
- PDaraei
- KWidjaja
- JWanagat
- TQAguiar Vallim
- AHFluitt
- SBensinger
- TLe
- CRadu
- JPWhitelegge
- SWBeaven
- PTontonoz
- AJLusis
- BWParks
- LVergnes
- KReue
- HSingh
- JCBopassa
- LToro
- EStefani
- MJWatt
- SSchenk
- TAkerstrom
- MKelly
- BKPedersen
- SCHewitt
- KSKorach
- ALHevener
-
- ABRubenstein
- GRSmith
- URaue
- GBegue
- KMinchev
- FRuf-Zamojski
- VDNair
- XWang
- LZhou
- EZaslavsky
- TATrappe
- STrappe
- SCSealfon
-
- MMSeldin
- GWWong
-
- MMSeldin
- SKoplev
- PRajbhandari
- LVergnes
- GMRosenberg
- YMeng
- CPan
- TMNPhuong
- RGharakhanian
- NChe
- SMäkinen
- DMShih
- MCivelek
- BWParks
- EDKim
- FNorheim
- KChella Krishnan
- YHasin-Brumshtein
- MMehrabian
- MLaakso
- CADrevon
- HAKoistinen
- PTontonoz
- KReue
- RMCantor
- JLMBjörkegren
- AJLusis
-
- MMSeldin
- AJLusis
-
- MSeldin
- XYang
- AJLusis
-
- MCKSeverinsen
- BKPedersen
-
- The Alliance of Genome Resources Consortium
-
- The UniProt Consortium
-
- LMVelez
- MMSeldin
-
- TWu
- EHu
- SXu
- MChen
- PGuo
- ZDai
- TFeng
- LZhou
- WTang
- LZhan
- XFu
- SLiu
- XBo
- GYu
-
- TYokota
- JMcCourt
- FMa
- SRen
- SLi
- T-HKim
- YZKurmangaliyev
- RNasiri
- SAhadian
- TNguyen
- XHMTan
- YZhou
- RWu
- ARodriguez
- WCohn
- YWang
- JWhitelegge
- SRyazantsev
- AKhademhosseini
- MATeitell
- P-YChiou
- DEBirk
- ACRowat
- RHCrosbie
- MPellegrini
- MSeldin
- AJLusis
- ADeb
-
- TZore
- MPalafox
- KReue